Como ya se ha especificado, una enfermedad rara o extraña, es aquella que se presenta en 1 de cada 10.000 personas, generalmente difíciles de tratar precisamente por su naturaleza y complejidad, aunque en muchas ocasiones los pacientes pueden llevar una vida normal dentro de lo que cabe. En este sentido, el síndrome Angelman es una rara enfermedad que tiene su origen como consecuencia de una mutación en el cromosoma 15, más precisamente en el gen UBE3A.
En condiciones normales, las personas heredan una copia del gen de cada uno de los padres, mismas que se activan en muchas de las áreas del cuerpo humano. Cuando solo una copia de este gen esta activo en determinadas áreas del cerebro, se tiene en consecuencia el síndrome Angelman, del cual no se conocen factores de riesgo, sin embargo en algunos casos un historial familiar puede incrementar las probabilidades de que un bebe padezca esta enfermedad.
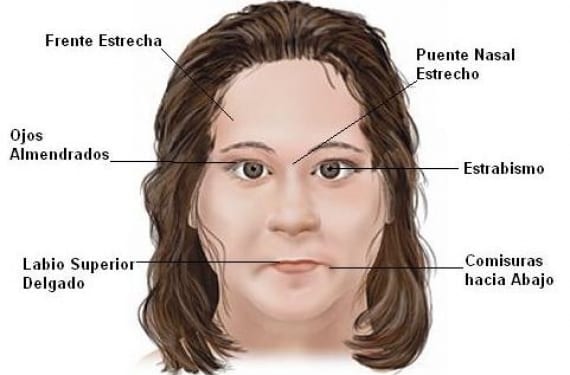
Los síntomas y características que se suelen presentar en los niños con síndrome Angelman, no necesariamente aplican para todos, por lo que pueden variar de un paciente a otro, sin embargo podemos identificar algunos comportamientos como por ejemplo:
- Agitar la mano o el pie con los brazos en el aire
- Tener movimientos corporales bruscos
- Poca o nula intervención
- Alto grado de hiperactividad
- Problemas de alimentación, particularmente en la etapa de la infancia
- Problemas de sueño
- Retraso en el desarrollo motor
- Risa frecuente en cualquier momento, incluso los menos apropiados
- Estrabismo
Finalmente y como mencionábamos al inicio, las personas que sufren del síndrome de Angelman pueden llevar una vida casi normal, sin embargo los adultos no suelen ser autosufientes para vivir por su cuenta, pero pueden aprender las tareas básicas del hogar e incluso es posible que algunas personas puedan conseguir un trabajo en el que estén supervisados directamente.
Más Información – Test de sangre para detectar el síndrome de Down
Fuente – childrenshospital.org
Está bien en cuanto a rasgos faciales. Escasa información, casi nula sobre otros aspectos.
Es posible que todos los rasgos no sean necesarios para diagnosticar este sìndrome? tengo una hija con 17 años y después de 6 años he dado con este síndrome, tiene de lección en el cromosoma 15, y por lo que estoy leyendo hay síntomas que parecen ser, donde me podría entera???